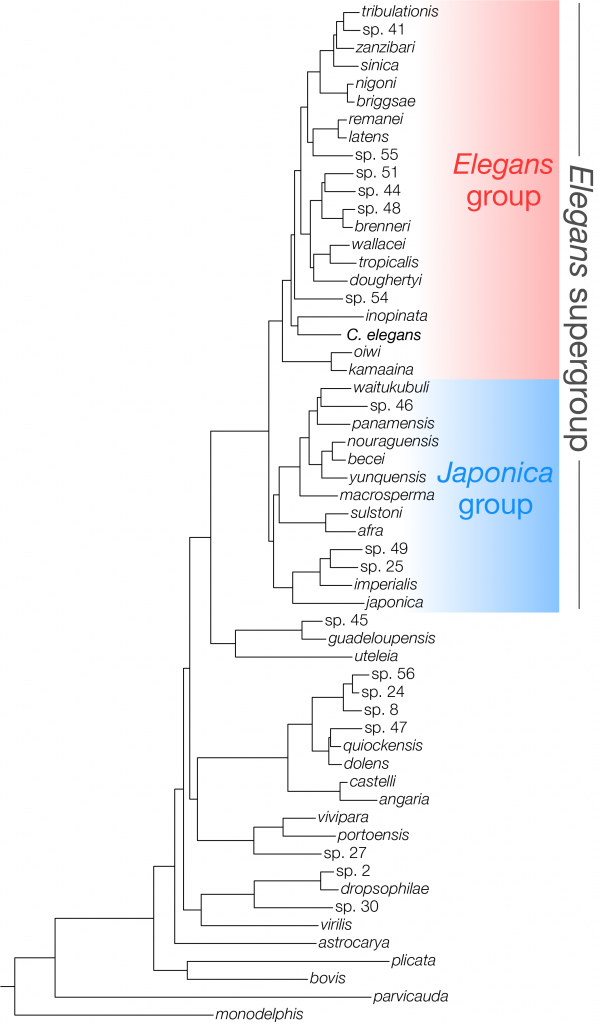
Below is the most recent Caenorhabditis phylogeny including 57 species. This tree was inferred using IQ-TREE using a supermatrix containing 982 single-copy orthologues (BUSCO nematode genes). It was rooted on two outgroup taxa, Diploscapter coronatus and Diploscapter pachys (not shown).
Note: this tree is preliminary and unpublished. For the most recent published phylogeny, see our paper in Current Biology.